-
 Australia
Australia
-
 Austria
Austria
-
 Belgium
Belgium
-
 Brazil
Brazil
-
 Canada
Canada
-
 China
China
-
 Czech Republic
Czech Republic
-
 Denmark
Denmark
-
 Finland
Finland
-
 France
France
-
 Germany
Germany
-
 Greece
Greece
-
 Hong Kong
Hong Kong
-
 Hungary
Hungary
-
 Iceland
Iceland
-
 India
India
-
 Ireland
Ireland
-
 Israel
Israel
-
 Italy
Italy
-
 Japan
Japan
-
 Korea
Korea
-
 Luxembourg
Luxembourg
-
 Malaysia
Malaysia
-
 Netherlands
Netherlands
-
 New Zealand
New Zealand
-
 Norway
Norway
-
 Poland
Poland
-
 Qatar
Qatar
-
 Romania
Romania
-
 Saudi Arabia
Saudi Arabia
-
 Singapore
Singapore
-
 Spain
Spain
-
 Sweden
Sweden
-
 Switzerland
Switzerland
-
 Taiwan
Taiwan
-
 Turkey
Turkey
-
 United Kingdom
United Kingdom
-
 United States
United States
research use only
NU6027 CDK inhibitor
Cat.No.S7114
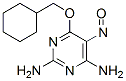
Chemical Structure
Molecular Weight: 251.28
Quality Control
Solubility
|
In vitro |
DMSO
: 50 mg/mL
(198.98 mM)
Ethanol : 3 mg/mL Water : Insoluble |
Molarity Calculator
|
In vivo |
|||||
In vivo Formulation Calculator (Clear solution)
Step 1: Enter information below (Recommended: An additional animal making an allowance for loss during the experiment)
Step 2: Enter the in vivo formulation (This is only the calculator, not formulation. Please contact us first if there is no in vivo formulation at the solubility Section.)
Calculation results:
Working concentration: mg/ml;
Method for preparing DMSO master liquid: mg drug pre-dissolved in μL DMSO ( Master liquid concentration mg/mL, Please contact us first if the concentration exceeds the DMSO solubility of the batch of drug. )
Method for preparing in vivo formulation: Take μL DMSO master liquid, next addμL PEG300, mix and clarify, next addμL Tween 80, mix and clarify, next add μL ddH2O, mix and clarify.
Method for preparing in vivo formulation: Take μL DMSO master liquid, next add μL Corn oil, mix and clarify.
Note: 1. Please make sure the liquid is clear before adding the next solvent.
2. Be sure to add the solvent(s) in order. You must ensure that the solution obtained, in the previous addition, is a clear solution before proceeding to add the next solvent. Physical methods such
as vortex, ultrasound or hot water bath can be used to aid dissolving.
Chemical Information, Storage & Stability
| Molecular Weight | 251.28 | Formula | C11H17N5O2 |
Storage (From the date of receipt) | |
|---|---|---|---|---|---|
| CAS No. | 220036-08-8 | Download SDF | Storage of Stock Solutions |
|
|
| Synonyms | N/A | Smiles | C1CCC(CC1)COC2=NC(=NC(=C2N=O)N)N | ||
Mechanism of Action
| Features |
A more potent inhibitor of cdk1 and cdk2 than NU2058.
|
|---|---|
| Targets/IC50/Ki |
ATR
0.4 μM(Ki)
CDK2
1.3 μM(Ki)
DNA-PK
2.2 μM(Ki)
CDK1
2.5 μM(Ki)
|
| In vitro |
NU6027 is soaked into crystals of monomeric CDK2 and the structure refined to a resolution of 1.85 Å. This compound (100μM) inhibits growth of human tumor cells with mean GI50 of 10 μM. It causes a reduction in the number of cells in S-phase but not G1 or G2/M in MCF7 cells. This chemical is a potent inhibitor of cellular ATR activity with IC50 of 6.7 μM in MCF7 cells and 2.8 μM in GM847KD cells, and enhances hydroxyurea and cisplatin cytotoxicity in an ATR-dependent manner. This inhibitor (10 μM) inhibits CDK2-mediated pRbT821 by 42% and pCHK1S345 by 70%. It significantly potentiates sensitivity of cisplatin (1.4-fold at 4 μM and 8.7-fold at 10 μM), doxorubicin (1.3-fold at 4 μM and 2.5-fold at 10 μM), camptothecin (1.4-fold at 4 μM and 2-fold at 10 μM) and hydroxyurea (1.8-fold at 4 μM) aganist MCF7 cells. It also potentiates 2Gy IR in a concentration-dependent manner and the cytotoxicity of camptothecin and temozolomide (a DNA methylating agent) at concentrations above and below their LC50. This compound (10 μM) attenuates G2/M arrest following DNA damage, inhibits RAD51 focus formation and increases the cytotoxicity of the major classes of DNA-damaging anticancer cytotoxic therapy but not the antimitotic, paclitaxel in MCF7 cells. It (4 μM) is synthetically lethal when DNA single-strand break repair is impaired either through poly(ADP-ribose) polymerase (PARP) inhibition or defects in XRCC1 in MCF7 cells. This chemical (4 μM) increases the proportion of cell in early apoptosis to 7.5% after 48 hours treatment in EM-C11 cells compared to 1.73% in untreated cells. This treatment (10 μM) reduces survival in XRCC1 deficient OVCAR-4 cells compared to proficient cells. It enhances cytotoxicity of cisplatin in XRCC1 deficient OVCAR-3 cells compared to XRCC1 proficient cells. This compound enhances Cisplatin induced DSB accumulation in XRCC1 deficient OVCAR-3 cells.
|
| Kinase Assay |
Enzyme Inhibition Studies
|
|
Inhibition of cyclin B1/CDK1 is assayed using enzyme prepared from starfish oocytes. Inhibition of cyclinA3/CDK2 is determined using a similar assay and an assay buffer comprised of 50 mM Tris-HCl pH 7.5 containing 5 mM MgCl2. The final ATP concentration in both CDK assays is 12.5 μM, and the IC50 concentration for this compound is the concentration required to inhibit enzyme activity by 50% under the assay conditions used. To determine the Km for ATP for cyclin B1/CDK1 and cyclin A3/CDK2, and Ki values for this chemical, assays are performed in the absence of this chemical and at two fixed concentrations of this compound (5 μM and 10 μM), with ATP concentrations ranging from 6.25 μM to 800 μM. Data are fitted to the Michaelis−Menten equation using unweighted nonlinear least squares regression.
|
References |
|
Tech Support
Tel: +1-832-582-8158 Ext:3
If you have any other enquiries, please leave a message.
Signaling Pathway Map
Products are for research use only. Not for human use. We do not sell to patients.
©Copyright 2013 Selleck Chemicals. All Rights Reserved.